
Aubrey West, 2016
Cystic fibrosis (CF) is one of a growing number of human diseases caused by inherited mutations that disrupt protein folding. It is caused by dysfunction of the Cystic Fibrosis Transmembrane conductance Regulator (CFTR), a cAMP-regulated ion channel that resides in the apical membrane of epithelial cells. CFTR dysfunction can occur by defects in protein synthesis, folding, intracellular trafficking, channel gating, chloride conductance, or plasma membrane stability. In each case, loss of CFTR results in abnormalities of water, chloride, and/or bicarbonate transport that lead to dysfunction of target tissues including: pancreatic insufficiency, increased sweat chloride, intestinal obstruction, and most importantly, chronic pulmonary infection, inflammation, and ultimately death due to respiratory failure.
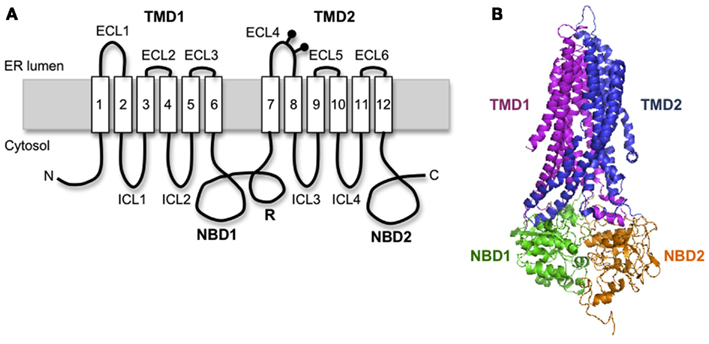
Cystic fibrosis transmembrane conductance regulator is a 1480 amino acid polytopic glycoprotein in the ABC transporter family (ABCC7) that contains two six-spanning transmembrane (TM) domains (TMD1 and TMD2) that form the channel pore, two cytosolic nucleotide binding domains (NBD1 and NBD2) that drive channel gating, and an intrinsically unstructured regulatory (R) domain that controls channel activity via PKA-mediated phosphorylation
Mechanisms of CFTR folding at the endoplasmic reticulum
| CFTR
NBD1 (Nucleotide Binding Domain 1) |
Page 1 |
| 14-3-3
and R Domain, Human NBD1 with Phe508 |
Page 2 |
| NBD1 with F580 Mutation and bound ATP | Page 3 |
| Human
CFTR NBD1, delta F508 with three solubilizing mutations |
Page 4 |
| CFTR
NBD2 (Nucleotide Binding Domain 2) |
Page 5 |
| CFTR Cif | Page 6 |